|
大家好: |
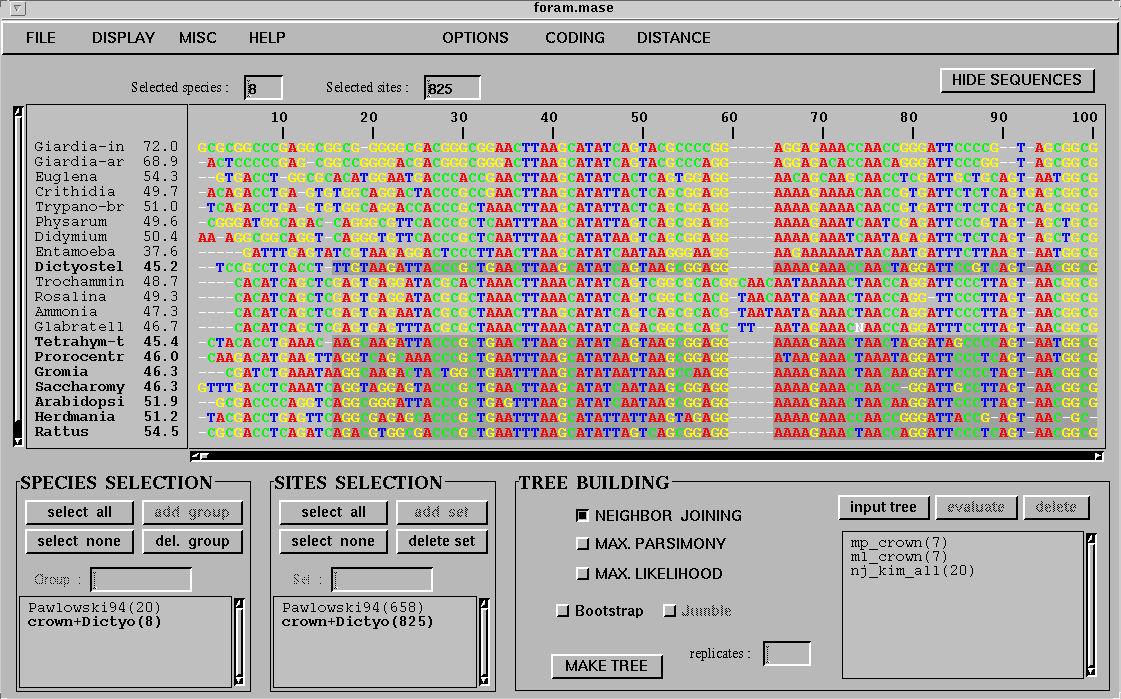
几个进化树分析及其相关软件的使用和应用范围
时间:2005-06-17 16:21来源:Biolover 作者:bioguider 点击:
1685次
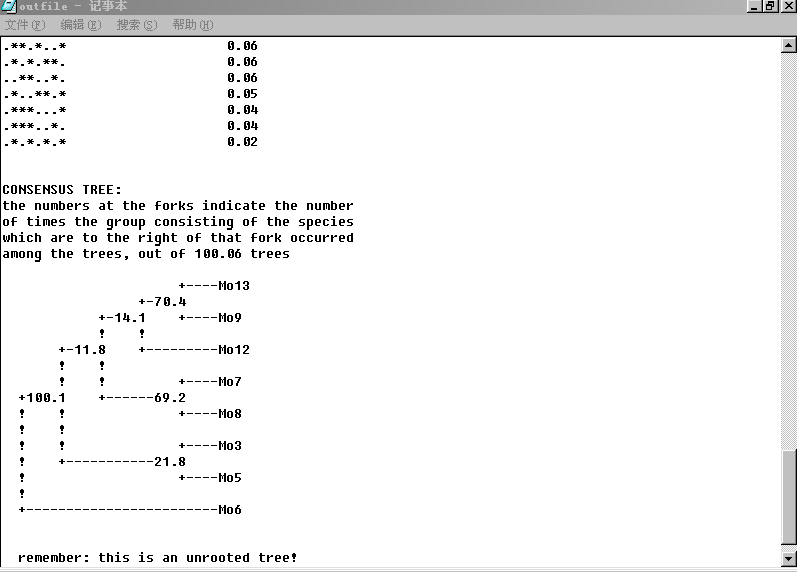
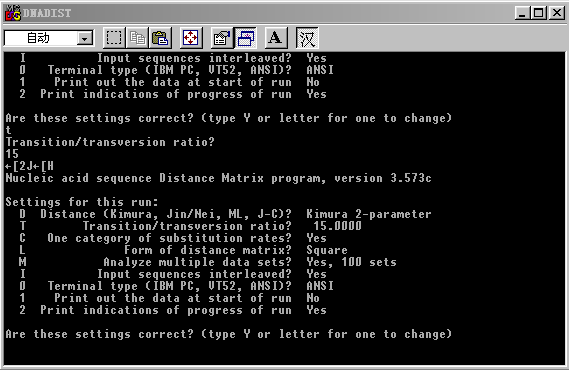
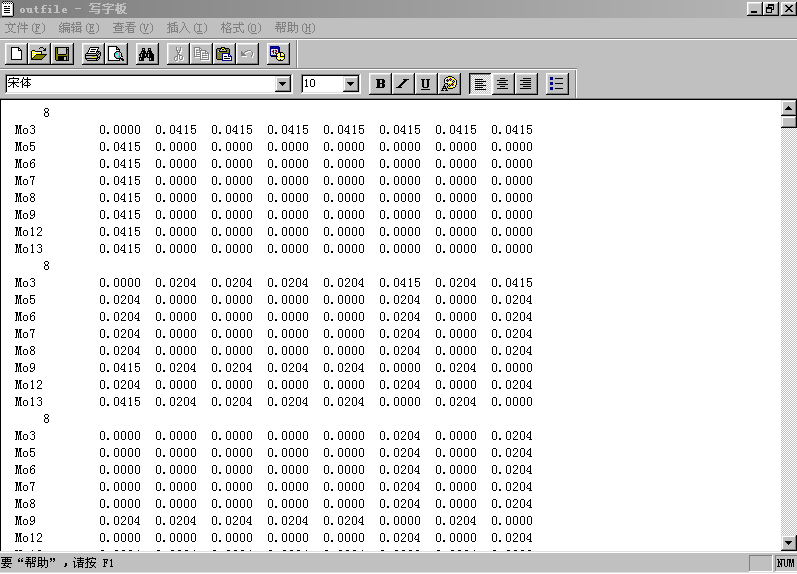
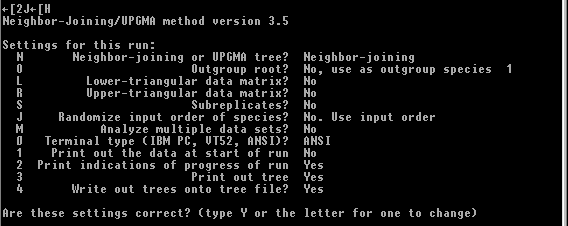
顶一下
(11)
100%
踩一下
(0)
0%
------分隔线----------------------------
- 上一篇:常用的统计计量数学软件(链接)
- 下一篇:SPSS 13版新增功能介绍
- 发表评论
-
- 最新评论 进入详细评论页>>
- 特别推荐
-

- 推荐内容
-
- Origin使用的一些经验
以下为本人自己使用Origin的一些经验,有一些虽然都是小问题,...
- SPSS中国落土生根 中国数据挖掘市场
随着中国管理类软件市场的蓬勃发展,数据仓库(DW),商务智...
- 网上免费统计资源
一、医学统计学教学 (一)医学统计学 课件名称: 回归与相关...
- 统计软件SPSS教程
...
- SCA 7 时间序列分析软件
SCA 7 时间序列分析软件 SCA 软件操作容易,适合各样背景的模型...
- Ox Metrics 介绍
Ox Metrics 經濟計量 分析軟體 族软件包的核心是 GiveWin -提供用户...
- Origin使用的一些经验