投资要点
我国创新药市场环境持续改善,前景广阔。创新药上市回报丰厚,研发能力要求高。目前,大部分药企仍处于仿创阶段,但随着药品政策环境不断优化,鼓励创新信号频发,产业迎来重要发展机遇。国内具备研发能力的创新药企不断涌现,老牌药企也纷纷布局。其中,生物类新药发展迅猛,中国有望借此弯道超车。当前,国家药品集中采购(带量采购)已常态化,倒逼企业向创新转型;医保谈判加速创新药准入,2020年以来每年开展国家医保谈判,创新药上市后平均1-2年即可纳入医保,较过去4-5年一轮的调整大幅缩短。此外,药品上市许可持有人(MAH)制度全面落地,优化资源配置,激发研发活力。截至2025年,我国已有超过80个1类新药获批上市,其中生物药占比超过40%。
他山之石:国外创新药研发对我国具有重要借鉴意义。国外创新药政策环境良好,相关机构重视专利保护,并采用多种优先审评模式加快新药审批,例如美国食品药品监督管理局(FDA)的突破性疗法认定、优先审评、快速通道和加速批准,欧洲药品管理局(EMA)的加速审评程序,日本药品医疗器械综合机构(PMDA)的优先审评程序。日本医药工业通过创新+并购重组+国际化,助力武田制药、第一三共等企业在不利的政策和市场环境中逆势生长。在相似产业环境下,日本经验对我国有重要借鉴价值。近年来,我国创新药企的国际化步伐加快,2023年信达生物的信迪利单抗、百济神州的替雷利珠单抗等PD-1抑制剂成功获得FDA批准,标志着中国创新药出海取得突破。
中国创新药企发展路径及估值探讨。1.0模式:老牌大型药企由仿到创,逐步布局创新药领域,如恒瑞医药、石药集团等,目前均有多个创新药产品线,恒瑞医药的卡瑞利珠单抗、阿帕替尼等已成为重磅品种。2.0模式:生而创新的小型研发企业,扎根创新药谱写新奇迹,如百济神州、信达生物、君实生物等,在肿瘤免疫、靶向治疗等领域取得瞩目成绩。百济神州的泽布替尼是首个获FDA批准的国产创新药,2023年全球销售额突破10亿美元。3.0模式:参与国际合作的开放式创新药企,通过许可引进(licence-in)和许可输出(licence-out)模式加速发展。创新药早期无利润,传统市盈率(PE)估值方法难以适用,通常依靠内生技术进步驱动,估值通过判断新药潜在市场大小、计算未来销售的净现值(NPV)数据。目前,科创板第五套上市标准为未盈利创新药企提供融资渠道,促进产业资本循环。
创新药研发机遇与风险并存。机遇:一级资本持续关注,2020-2024年生物医药领域融资总额超过3000亿元,降低对研发成本的担忧。风险:靶点同质化严重,PD-1、CAR-T等热门赛道竞争白热化,me-too药物市场空间受限;创新药研发失败率高,仅有约10%的候选药物能最终上市;国际化面临海外监管和市场竞争挑战。同时,医保谈判降价压力较大,创新药上市后需快速放量才能实现商业价值。
相关投资标的。恒瑞医药:新药研发领军企业,创新产品线丰富,拥有多个1类创新药,卡瑞利珠单抗、阿帕替尼等持续放量。百济神州:全球化布局最完善的创新药企,泽布替尼、替雷利珠单抗海外销售强劲,营收快速增长。信达生物:信迪利单抗已纳入国家医保,在肺癌、肝癌等大适应症中持续拓展,生物类似药组合丰富。君实生物:特瑞普利单抗获FDA批准,国际化取得里程碑,同时布局新冠中和抗体、BTLA抑制剂等前沿靶点。
风险提示:创新药行业鼓励政策推进不及预期;创新药研发失败风险;创新药上市后安全性风险;医保和招标政策变更对创新药带来的风险;药品集中采购对仿制药业务的冲击。
正文
1、我国创新药发展前景光明
1.1 药政巨变,蕴藏创新机遇
国家政策和财政投入在我国创新药物研发中起着决定性作用,影响创新药研发的政策包括:宏观经济和产业政策、科技政策、注册监管政策、医保支付政策、财税金融政策以及采购政策等。在早期临床前研究阶段,国家通过重大新药创制专项,以及国家自然科学基金的研究项目和人才项目等进行有计划的财政投入,高校及科研院所作为研发主体,具有一定市场前景后企业介入继续开发;新药进入临床阶段后,国家药品监督管理局(NMPA)制定相关法规进行指导;创新药物上市后,面临招标、入院、入医保三道关卡。近年来,国家医保局推动药品集中带量采购和医保谈判,加速创新药市场准入。截至2024年,已有超过200个创新药通过谈判纳入国家医保目录。国家政策和政府财政投入贯穿创新药物研发的各个阶段,推动我国创新药产业的发展。
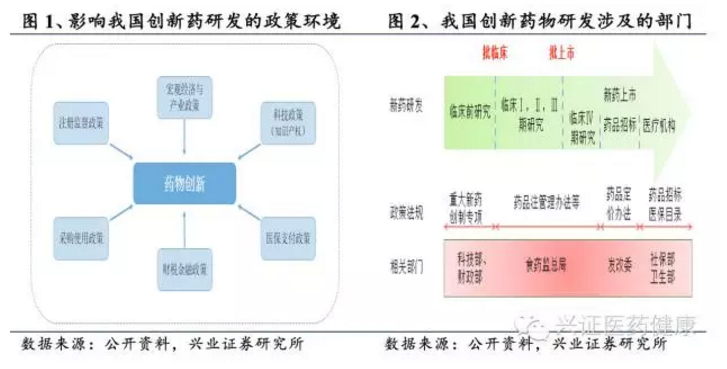
目前,我国新药研发及注册相关法律法规体系不断完善,包括《药品管理法》(2019年修订)、《药品注册管理办法》(2020年修订)、《专利法》(2021年修订)等。其中,《药品注册管理办法》设立突破性治疗药物、附条件批准、优先审评审批、特别审批四个加快通道,与国际接轨。2015年以来,药品政策法规密集出台,提高药品研发壁垒和质量的同时,也促使医药研发和合同研究组织(CRO)行业更加规范化和集中化,促进行业优胜劣汰。

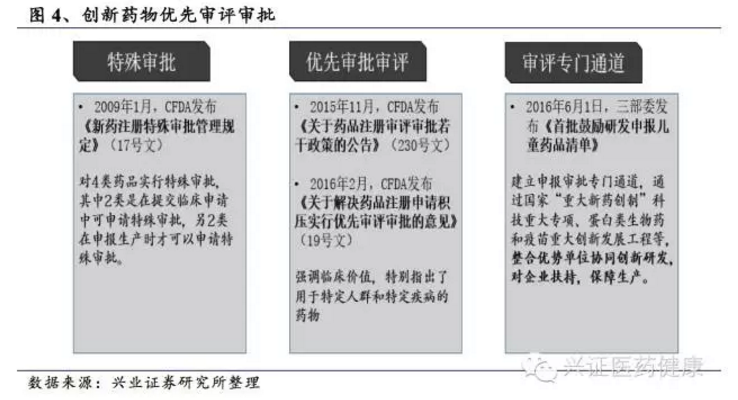
创新药审批加速
2015年11月,原国家食品药品监督管理总局(CFDA)发布《关于药品注册审评审批若干政策的公告》,提出对新药的临床试验申请实行一次性批准,不再采取分期申报、分期审评审批的方式,仿照FDA审批制度优化审批流程,有利于加快企业研发进度,加快新药上市,降低企业时间成本,延长产品生命周期;对重大疾病的创新药等8类药品实行单独排队,加快审评审批。2020年《药品注册管理办法》实施后,进一步明确突破性治疗药物程序,对具有重大临床价值的创新药给予全程指导。在优先审评审批方面,截至2024年,NMPA已将超过400个注册申请纳入优先审评,涵盖抗肿瘤、罕见病等重点领域。审评审批周期大幅缩短,临床申请平均审评时间由过去的12-18个月缩短至60个工作日以内。
化药注册分类改革重新定义创新药的概念
为鼓励新药创制,严格审评审批,提高药品质量,促进产业升级,原CFDA于2016年3月发布《化学药品注册分类改革工作方案》,对化学药品注册分类进行改革。新版方案将新药定义从“中国新”提升至“全球新”,新药必须是境内外均未上市的药品,并进一步分为1类新药(创新药)和2类新药(改良型新药)。新注册分类1指含有新的结构明确的、具有药理作用的化合物,且具有临床价值的原料药及其制剂;新注册分类2改良型新药强调“优效性”,即相较于被改良的药品,具备明显临床优势。此后,2017年NMPA加入国际人用药品注册技术协调会(ICH),推动药品审评标准与国际接轨。2021年《化学药品注册分类及申报资料要求》进一步细化,明确1类新药包括境内外均未上市的原创新药,涵盖化学药、治疗用生物制品和预防用生物制品。临床价值评估在新药研发中越来越重要,首仿品种价值被削弱,创新药成为研发主流。

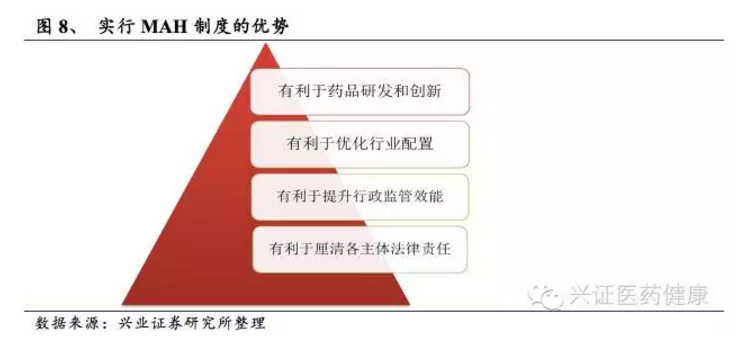
MAH制度优化资源配置,调动研发企业积极性
药品上市许可持有人(MAH)制度将药物上市许可与生产许可分离的管理模式。上市许可和生产许可相互独立,上市许可持有人可将产品委托给不同生产商生产,药品安全性、有效性和质量可控性由上市许可人对公众负责。MAH制度是国际较通行的药品上市审批制度。2016年国务院办公厅印发《药品上市许可持有人制度试点方案》,在北京、天津、河北、上海等10省(市)开展试点。2019年新修订《药品管理法》正式确立MAH制度在全国实施。MAH制度很大程度上调动了研发者的积极性,促进药品创新;药物申报对药厂的依赖度显著降低,利好创新性中小企业;同时优化资源配置,抑制低水平重复建设,落实企业主体责任。截至2024年,全国已有超过3000个药品批准文号通过MAH制度实施委托生产,培育出一批轻资产创新型公司。
总体来看,这些政策的出发点是与国际接轨,鼓励创新。药监部门把药品质量向国际先进水平看齐,国内药品开发今后会更多考虑患者需求和临床应用,这也是国外药企研发的思路。很多有实力(有技术、经验、资金)的公司会有很大机会,以后的创新必须是有研发体系、有资金支持的企业才能玩的高投入游戏,小企业将逐步走向差异化或细分领域,带来行业集中度提升。我们认为,在监管部门的新政策引导下,企业的研发战略、市场战略必将迎来大转型,创新药和制剂出口将受益于政策利好,迎来新的发展机遇。
1.2 我国新药研发环境显著改善,创新药研发如火如荼
创新药物是医药领域利润最丰厚、最能体现技术含量的部分,未来这一领域最能维持高利润水平。从长远来看,创新药物的研发能力仍然是企业的核心竞争力。新药研发要经历化合物筛选、临床前研究、临床试验、注册申报等过程,5000-10000个候选化合物才能有一个药物最终上市,一个创新药研发周期耗时长达10年,平均研发费用达数十亿美元。因此,创新药研发需要巨大的资金投入和强大的研发实力作为支撑。
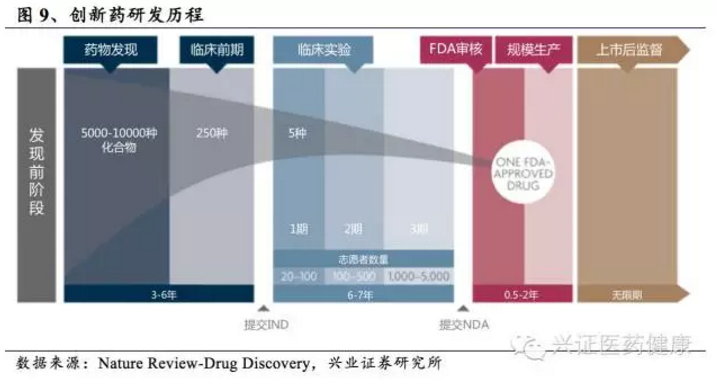
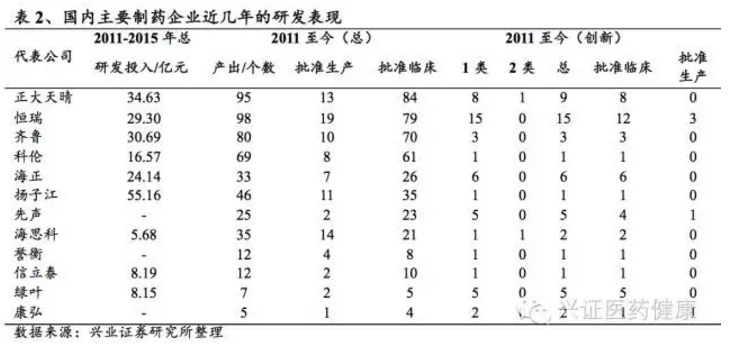
以国外创新药巨头强生为例,2002-2012年间共上市新药13个,累计研发投入676.2亿美元,平均每个新药研发成本为52.0亿美元;国内创新药研发成本相对较低,以恒瑞医药的艾瑞昔布为例,研发成本约1-2亿元。尽管如此,由国内化学药上市公司的盈利水平和研发投入情况来看,也只有中国生物制药、恒瑞医药、百济神州等少数企业才有能力持续投入大规模研发费用。2023年,百济神州研发投入超过128亿元人民币,位居中国药企首位。
根据美国FDA的规定,创新药物包括四类:药品中含有新化学实体(NCE)作为活性成分;药品含已有活性成分但该成分在美国从未作为医学用过;药品先前已被FDA批准上市,但现在建议新用法、新适应症;药品先前已被批准上市,但现在建议新剂型、给药途径或其他重要条件不同于先前获批的药品。目前创新药物的主要研发方向有:1)创制新颖分子结构类型(NCE/NME)——突破性创新药物研究开发。2)创制me-too新药——模仿性创新药物研究开发。3)已知药物的进一步研究开发——延伸性创新药物研究开发。4)现有药物的药剂学研究开发——发展制剂新产品。5)应用现代新技术对老产品生产工艺进行重大革新和技术改造。
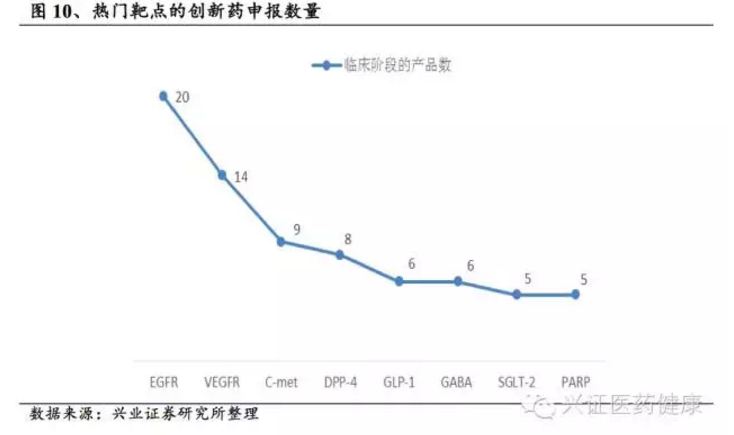
我们认为中国的创新药研发可分四步走:首先从“仿制”转型为“创新”,即研制me-too药物,如恒瑞医药的艾瑞昔布;第二步是me-better,围绕原NCE结构进行二次创新,如贝达药业的埃克替尼、恒瑞医药的阿帕替尼;第三步是best-in-class(BIC),是me-better中的佼佼者,例如辉瑞的万艾可;第四步是first-in-class(FIC)药物,即全新化合物且靶点全新,研发风险极高,但成功者众多,如获得诺贝尔奖的青蒿素。我国当前创新层次主要以仿制为主到仿创结合阶段,仿制药占比仍高达80%以上,上市药物多为me-too药物,新药市场被国际大公司产品垄断,缺乏first-in-class药物。对国内企业而言,结合国内临床需求,在国际新药基础上开发药效和安全性相似的me-too或更好的me-better药物,仍是目前比较切实可行的创新路径,也是国内创新药研发的主流方向。
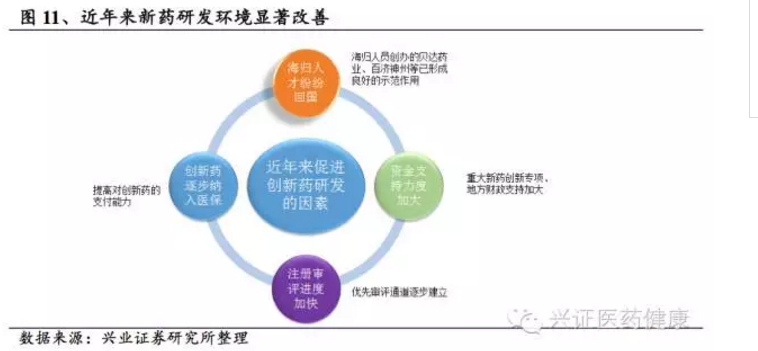
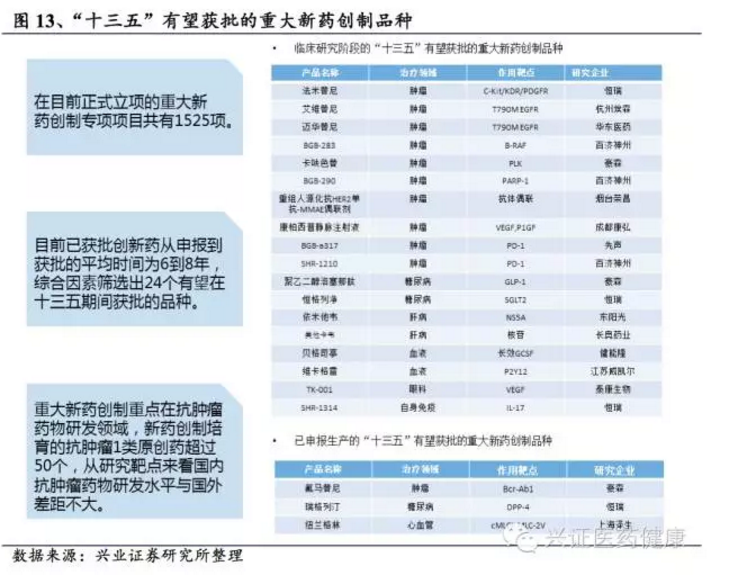
近年来,我国新药研发环境显著改善,大量海归人员创业,百济神州、贝达药业等企业的成功形成良好示范效应;在鼓励创新、开辟绿色通道等政策推动下,创新药研发和审评周期进一步缩短。如泽布替尼从临床到获批仅用5年,埃克替尼申报上市仅用10个月,有效延长产品生命周期;1类新药逐步纳入各地医保也很大程度上鼓励了新药研发。截至2024年,我国在研创新药数量超过5000个,临床试验数量大幅增长,其中约30%为生物药。
目前,国内确实有一批企业在创新药方面进行了布局,如恒瑞医药、石药集团、百济神州、君实生物、信达生物、贝达药业等,国内创新药研发热情持续升温,取得多个里程碑式成果,多个新药正在申报或获批中。
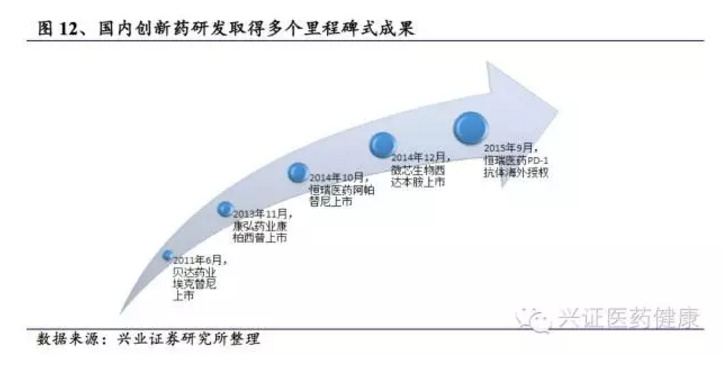

“十三五”以来,针对重大疾病围绕产业链部署研发链,获批1类新药超过80个,建成各类平台近300个,突破核心关键技术50余项,获得国家奖项50余项,国家经费投入超过70亿元,产生直接经济效益数千亿元,出口产值达数十亿美元。生物疫苗研发水平位居世界前列,化学药物创新研究实现与国际同步,中药产业形成全球化发展趋势,促进我国医药科技由仿制向创制、医药产业由大国向强国转变。
1.3 生物技术将成为创新药物研发新引擎
全球范围内,化学药仍是创新药的主要来源,但生物类药物增长迅速。从1986年FDA批准第一个治疗性单抗OKT-3开始,1986-2014年间共125种治疗性生物药获批,占所有新分子实体(NME)的15%;近十年(2005-2014)共有55种生物药获批,占NME的20%(共269种)。2014年,化学合成小分子物质仍是候选药物的主要来源,占比55.4%,而生物药呈现快速增长态势,在研品种达3123种,增幅13.5%,占在研药物总量的27.6%。到2024年,生物药在研品种已超过6000种,占比超过35%,其中单克隆抗体、细胞疗法、基因疗法成为热点。
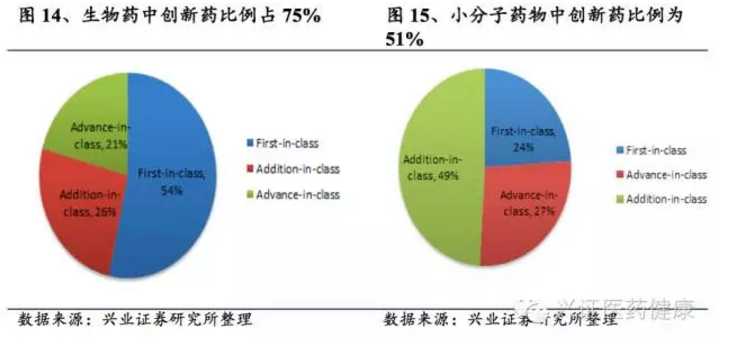
在1986-2014年获批的生物药中,first-in-class占54%,advance-in-class(药效优于已获批药物,相当于me-better)占21%,仅26%被评为addition-to-class(与现有药物相比无显著提高);而小分子药物中前两类比例分别为24%和27%,非创新药比例达49%。可见,与小分子药物相比,生物药更易成为创新型药物,且更多聚焦于罕见病领域,47%的生物药被定为孤儿药,而小分子中仅21%。
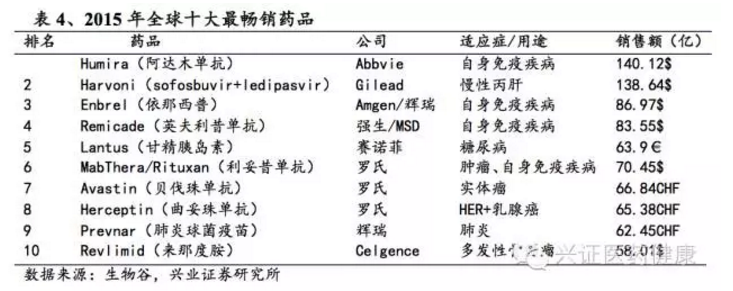
2015年全球最畅销十大药物中,生物药占8个。到2024年,全球药品销售额前十名中生物药仍占据主导,包括阿达木单抗、帕博利珠单抗、纳武利尤单抗等。目前,国际上主流生物技术药物产品与研发重点以单克隆抗体为主,同时细胞治疗(如CAR-T、TCR-T)、基因治疗、抗体药物偶联物(ADC)等新兴领域快速发展。未来几年,随着化药市场成熟,生物药仍处成长期,其领域创新药研发发展前景被看好。
我国生物技术药物产品包括干扰素系列、促红细胞生成素、集落刺激因子系列、肿瘤坏死因子、胰岛素和生长激素等。近年来,疫苗得到进一步发展,尤其治疗性乙肝疫苗、生长激素、艾滋病疫苗,以及类似H1N1等突发流行性因素产生的疫苗需求持续扩大。与癌症治疗相关的重组蛋白、单克隆抗体等基因工程,以及胰岛素、检测试剂等领域,发展潜力巨大,也是未来几年增长较快的生物医药投资热点。在肿瘤免疫治疗方面,PD-1/PD-L1抗体药物领域已有超过20家企业获得上市批准或进入临床,竞争激烈但市场空间巨大。
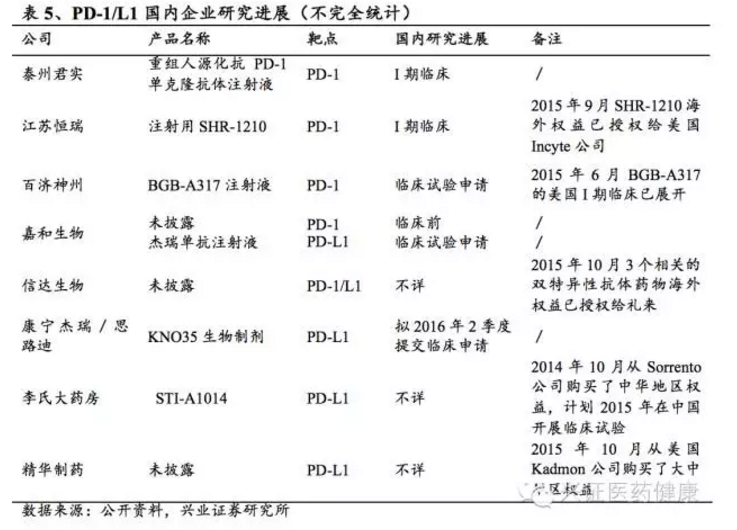
从大型药企(Big Pharma)角度看,小分子药物研发始终是重点,但生物药关注度不断提高;中等制药企业(Midcap Pharma)对生物药关注度也在增加,不过较缓慢;生物技术公司(Biotech)大部分精力都在生物药上,包括单抗和各种治疗性蛋白质。这些小型创新型生物技术公司通常有独特竞争优势,能够实现跨越式发展,如我国的百济神州、信达生物、微芯生物、再鼎医药等都属此类。
1.4 我国创新药研发呈现和国际合作趋势
对于制药企业而言,没有持续不断的好产品就走不远。新药研发投入越来越大,投入产出比却越来越少,所以不少大型药企一方面通过并购取得新产品,另一方面,通过项目许可(licence-in/out)加速开放式创新研发模式流行。近年来,国内创新药企与海外药企合作事件频发,包括将研发到临床二期或三期的新药海外权益转让给海外公司的licence-out模式,以及引进海外二期或三期药物的licence-in模式,逐步参与国际化创新分工。
Licence-out:走出去
2024年全球药企市值前十名中,强生、辉瑞、默沙东等市值均超过2000亿美元,而国内医药上市公司市值最高的恒瑞医药(截至2024年底)约3000亿元人民币。欧美Big Pharma大多经历百年沧桑才有今日之巨,以仿制药起家的中国医药企业无论从经验还是规模上都难以匹敌。从细分领域切入,依靠合作和授权走向全球化销售,可能是更现实的方式。2022-2024年,中国创新药海外授权交易数量快速增长,2023年超过50笔,总金额超过400亿美元,其中百济神州的替雷利珠单抗、科伦博泰的ADC、和黄医药的呋喹替尼等产品成功授权给国外药企。通过licence-out,可以利用海外公司相对丰富的经验组织海外临床试验,降低海外临床难度;海外推广、上市后再评价等工作依赖海外公司支撑,为新药全球上市和后续研究提供保障;同时,国内企业可学习管理研发经验,培育国际化人才,增强综合实力。
Licence-in:引进来
近年来,我国一些研发效率高的小型药企引进国外药企品种,挖掘国内市场潜力。合作模式中,国内企业大多获得中国地区独家转让权,全球范围转让较少。中国药物注册窗口相对收紧,新药在中国上市速度加快但与国际仍有差距,注册环节时间花费成为关键竞争指标。创新药物以国内企业身份申报新药,既能针对亚裔人群特点进行开发,又能加快审批上市速度。《“十四五”医药工业发展规划》明确提出支持创新药国际化,鼓励引进国外先进技术和产品。截至2024年,国内引进的创新药项目超过300个,涵盖肿瘤、自身免疫、罕见病等领域。
2、他山之石:国外创新药物研发对我国有借鉴意义
2.1 国外鼓励药物创新的政策环境
美国对创新药物研发采取的主要政策
美国是新药开发能力最强的国家,除企业自身利润追求、政府法规政策鼓励外,还与FDA在注册审批中的技术引导和帮助分不开。在专利保护方面,1984年《药品价格竞争和专利期恢复法》(Hatch-Waxman法案)允许药品专利获得长达5年的专利期延长。1994年《乌拉圭回合协议法》将专利保护期从批准日起17年变更为最早申报日起20年。2012年《食品药品监督管理局安全及创新法案》设立突破性疗法认定,加快审批;申请人可滚动提交资料和随时沟通交流。FDA的鼓励措施以临床应用为导向,主要针对治疗严重和危及生命疾病的新药、比已上市药品有显著改进的药品,设有四条特别通道:1)突破性疗法认定(Breakthrough Therapy):要求初步临床证据表明药物在治疗严重或危及生命疾病方面较现有疗法有实质性改进,可获得FDA全程指导、滚动审评和优先审评资格。2)优先审评(Priority Review):针对治疗、诊断或预防疾病上比已上市药品有显著改进的药品上市申请,审评时间为6个月(标准审评11个月)。3)快速通道(Fast Track):针对有潜力治疗严重和危及生命疾病的新药研发和审批,FDA早期介入,申请人可分阶段递交资料。4)加速批准(Accelerated Approval):允许基于替代终点或中间临床终点批准新药,批准后需进行确证性研究。2000年代以来,多个重磅抗癌药通过加速批准上市。
欧盟对创新药的专利保护及优先审评政策
在专利保护方面,欧盟2004年药品管理法修正版统一了数据保护时间:创新药享受8年注册信息保护期,之后继续享受2年市场独占期(“8+2+1”公式)。在两种情况下,创新药可延长1年注册信息保护:1)批准新适应症且有显著临床收益;2)药品从处方药转为非处方药(OTC)的数据保护。在优先审评方面,欧洲药品管理局(EMA)的加速评审程序主要针对具有重大公共卫生效益的医药产品,如甲型H1N1流感疫苗等。加速审评程序时限为150天(标准210天),要求申请人证明该产品具有重大公共卫生效益。
日本PMDA的优先审评程序
日本PMDA的优先审评程序适用于治疗严重疾病且优于现有治疗方法的药品或医疗器械。严重疾病指对生命有重大影响或病情进展不可逆、严重影响日常生活的疾病。优于现有疗法指目前无有效治疗、预防方法,或安全性、有效性、患者负担等方面优于现有疗法。产品需至少提供Ⅱb期临床结果。优先审评程序审评时间为6个月(常规12个月),并可优先安排与PMDA咨询。
2.2 美国重磅创新药物不断涌现,造就无数明星药企
2015年FDA共批准45个新药,创近10年新高。2015年获批的新药中,有33个新分子实体和12个生物制品,包括14个抗肿瘤药物、6个心血管药物、5个精神类药物和2个抗感染药物。2023年FDA批准了55个新药,其中21个为first-in-class。2024年全球药品销售额前十名中,默沙东的K药(帕博利珠单抗)以250亿美元位居榜首,百时美施贵宝的O药(纳武利尤单抗)超过100亿美元。吉利德索非布韦及其复方Harvoni曾在丙肝治疗领域创造销售记录,2020年后其地位被新一代直接抗病毒药物替代。百时美施贵宝在肿瘤免疫领域占据重要地位,其PD-1抑制剂纳武利尤单抗联合伊匹木单抗获批多个适应症。2022-2024年,ADC药物(如Enhertu、Padcev)和双特异性抗体(如blinatumomab)成为新的增长点,罗氏、阿斯利康、第一三共等企业表现突出。
2.3 创新+并购+国际化,助力日本创新药企腾飞
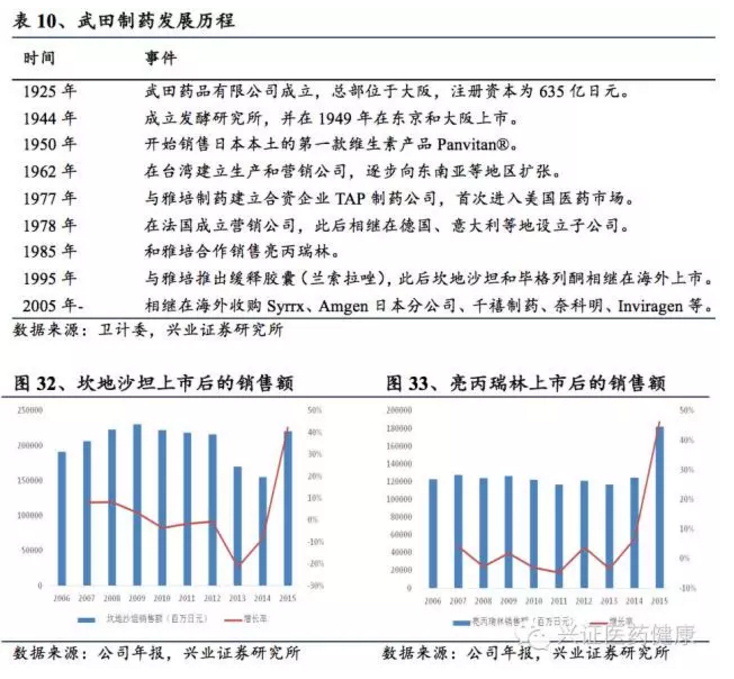
日本在上世纪70年代中期后,随着经济增速和财政收入下滑、医保覆盖率提升和人口老龄化带来的医保支出压力增大,控费压力直接影响医药工业运行。面对压力,日本医药工业企业探索三种发展路径:1)创新:通过新药“创新溢价”降低行政降价负面影响;2)并购重组:大型企业通过强强联合扩大市场份额;3)国际化:逐步进军欧美等海外规范市场。创新+并购重组+国际化驱动武田制药、第一三共、安斯泰来、大冢等企业逆势生长,使日本医药产业再度腾飞。武田制药从1781年传统中药销售起步,后转型为医药制造商,1978年在法国设立营销公司,逐步推出亮丙瑞林、兰索拉唑等创新药,2000年后收购Syrrx、千禧制药、奈科明、Inviragen等海外企业,并收购爱尔兰夏尔公司,成为全球十大药企之一。2020年以来,武田专注于罕见病、肿瘤和消化领域,并在基因疗法等领域布局。日本经验表明,创新叠加国际化和并购是实现跨越式发展的关键。
案例:武田制药
武田制药发展可追溯至1781年创始人武田从事传统中药销售。1895年转型为医药制造商,1914年成立研究部门,推出自有品牌。70年代后面对国内增速放缓,1978年在法国设立营销公司进军欧洲。随后推出亮丙瑞林、兰索拉唑等创新药,坎地沙坦和毕格列酮在欧美上市。2000年后开展全球扩张和兼并,收购Syrrx、千禧制药、奈科明、Inviragen等,扩充商业版图。武田的发展遵循“原料药→仿制药→创新药+国际化”路径,既减轻对国内市场的依赖,提高抗风险能力,又嫁接全球资源,推动更好研发。
2.4 负利润大市值,台湾鼓励创新药企发展
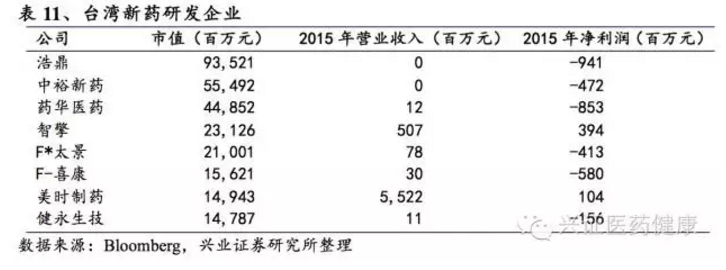
台湾创新药企发展分两阶段:第一阶段是90年代以前,国际大型生物医药厂商控制研发核心技术,台湾以中小企业为主无法承担巨额研发支出,创新效率低下;第二阶段是90年代中期以后,国际大型厂商倾向研发外包,加之台湾官方大力扶植生物技术产业,从直接介入研发转向辅助建立研发平台,出现大量小型企业专注委托外包研发或制造,传统药厂从生产专利过期的学名药转向研发创新药,形成研发新药、卖专利的发展模式。台湾政府持续出台利好政策,2011-2014年实施“生技医药国家型科技计划”,筛选出产业化潜力技术,其中“亮点计划”最受关注。政府支持未获利新药公司根据科技事业条例上市柜募资,台湾市值最大的生物医药公司中,无营收、负利润的公司如浩鼎(市值935亿台币)、中裕新药(555亿台币)等赫然在列。这种模式鼓励企业大胆投入创新,但风险较高,对大陆创新药生态有一定借鉴意义。