|
对于神经科学中分子生物学部分,特别是相关疾病方面的研究手段并不是很多。特别是对于多个因素的作用,现在的研究手段还比较稚嫩,对于DNA的初步大规模分型无疑是一种好的方法。AFFY用基因芯片来检测SNP达到了高通量的目的,非常适合于第一步的 筛查。 复杂DNA的大规模基因分型 Nature Biotechnology, Volume 21, Number 10, October, 2003
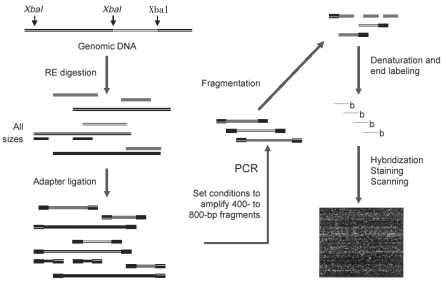
我们克服了当前的基因型技术中的2个重要瓶颈:位点特异性SNP 的扩增和位点特异性等位基因区别的要求 。我们设计了基因样本的准备方法,通过单核苷酸引物进行扩增,用DNA 芯片检测等位基因。基因芯片通过样品中的核酸分子同芯片上的互补性顺序进行的特异性杂交来确定大量的基因信息。 尽管目前可以在芯片上合成大于500,000探针序列,但是主要的挑战是如何将基因组DNA放在芯片上的同时得到有关样本的准确的等位基因信息。许多靶基因的特殊基因碱基对(复杂性)增加了交叉杂交以及非特异性信号的机率。因此,优先选择基因组的一部分(或者片段),在芯片上才可以得到有意义和特异性的信号。 进行基因组的筛选目前有几个已知的方法。共同的策略是使用限制性酶消化,随后进行接头连接,并用一个引物进行扩增。所采用的方法的不同在于降低复杂度的步骤。举个例子来说,代表差异性分析利用PCR选择性扩增长度至1kb的片段。这种扩增的片段长度多态性(AFLP)的方法是利用特异性引物去扩增基因。基因片段也可以通过凝胶选择片段大小来制备。TSC (The SNP Consortum) 利用这种方法确认了超过一百万SNPs。所有这些方法在制备片段化的基因方面都取得了进步,但是它们在区别等位基因方面仍有局限性,即在大规模的基因型确认方面仍有缺陷。 我们所建立的基因分型方法中满足3种标准。首先,它应该覆盖TSC公共数据库中的大量SNPs的数量。其次,为避免SNP特异性的引物,所得到的大量的SNPs是经过仔细选择。最后,为保证准确性必须是可被高度重复的。 为了利用大量已发现的SNP位点,我们致力于TSC和SNP发现中所采用的片段化方法,即用EcoRI, BglII和 XbaI 限制性酶进行消化, 接着选择了400- 800bp范围 的片段。我们通过优化扩增条件并且选择性扩增这种大小的片段来代替凝胶法分离片段大小。我们所采用的生物化学方法是通过PCR 或者FSP的方法来进行片段化选择,可见图1及本文下面的详细描述。 图1通过PCR进行片段选择(FSP)
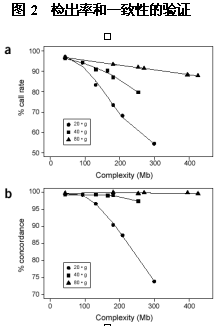
由FSP所产生的靶目标被标记后同芯片杂交。每个EcoRI, BglII 以及 XbaI 片段代表接近4×107 bp基因组DNA。典型的同类片段芯片杂交显示很强的信号密度,而相等量的人类全基因组DNA (3.2×109bp)芯片杂交信号密度就低得多。SNP通过等位基因特异性杂交, 并用针对高度复杂的样本的统计学分析得到。我们用108个个体验证这种统计算法。我们观察到与三种可能的基因型相关的基因簇(Cluster),一套保守算法确认了14,548高质量的SNPs。测定了38个样本检测的重复性和准确性(Methods),发现可以达到95.8%的平均检出率(检出的SNP总数除以总的检测数),同其他基因分型的方法相比有约99.1% 的一致性。 我们用检测了3种基因片段,每个片段接近43Mb,杂交不同的分别芯片。然后,我们通过产生一系列从43到425Mb复杂度增高的目标样本检测检出率及一致性。我们将这些样本同SNP芯片杂交,来检测XbaI 消化后的片段,并确定3种靶DNA量的检出率和一致性 ( 图2a,b)。随复杂度增加, 检出率和一致性均降低;然而,当靶片段总量低时这种效果则更明显。因此,有可能通过提高靶DNA的量对高度复杂的样本(>300Mb)进行基因分型,并达到99%以上的准确性。
我们用WGSA确定了来自于60个无关个体的DNA序列中的SNP等位基因频率。这些个体分别来自于3大人类种族:非裔美国人,高加索人以及亚洲人。主要的SNP位点在所有3种人群中是多态性的。这同含有多种族的验证组(包括这3种人种以及另外2种(当地美国人和墨西哥裔美国人))的预期是一致性的。在此分析中,非裔美国人、高加索人和亚洲人样本中分别有343,535和1,219个标记是单态性(也就是杂合性为0)。 我们这次研究的标记在非洲美国人、高加索人和亚洲人中的平均杂合性为0.348, 0.354和 0.322,表明大部分的SNP信息可用于种群研究。 我们用FST的统计算法,一种对2种种群间地理结构的评估, 对每个SNP进行计算。FST值在0到1之间变动;当种群间等位基因频率的差异增高时,FST值也随之增高。非洲美国人对高加索人,非洲美国人对亚洲人,高加索人对亚洲人的FST分别是0.061、0.094和 0.065,因此,大多数标记表明种群间频率差异小。这些值同以前报道的在不同样品系列中通过较少的位点计算出的FST值一致。非裔美国人和亚洲等位基因频率的比较比另两种比较得出的FST值总体来看要高一些。我们的研究显示了尽管大多数SNP在三种人群中表现较低的等位基因频率差异,有一组SNP其等位基因频率在一个种群中显著不同于另外2组。这些祖先-信息相关的标记物(AIMs),可用于MALD(map complex diseases using admixture-generated linkage disequillibrium)分析,定位多种复杂疾病。在非裔美国人对高加索人,非裔美国人对亚洲人和高加索人对亚洲人的比较中,分别有343,788和 374个 SNP位点其FST值大于0.4。 SNP为进化过程中出现的序列改变。为了确定哪些等位基因代表祖先状态(ancestral state) ,我们将黑猩猩和大猩猩的DNA样本进行基因分型。黑猩猩和大猩猩的DNA分别与人存在1.5%和2.1%的差别。我们曾用涵盖14,548个人SNP位点的人SNPs芯片对黑猩猩和大猩猩进行基因分型研究。我们对黑猩猩和大猩猩的基因型检出率分别为77.1%和71.8%(数据未提供)。几乎所有标记在两种大型猿中表型为纯和的:对黑猩猩为97.8%,大猩猩为97.7%,这同SNP进化的历史是一致的。我们设定祖先等位基因(ancestral alleles)只针对哪些在黑猩猩和大猩猩中都为纯和的SNP位点,在这两种物种中为同样的基因型。总共有8,386个这样的SNP位点。 (责任编辑:泉水) |
关于SNP与神经生物学起始研究的一种工具
时间:2006-03-22 22:14来源:本站原创 作者:hawkgo 点击:
431次
顶一下
(5)
100%
踩一下
(0)
0%
------分隔线----------------------------
- 上一篇:日研究发现鲑鱼鼻软骨对炎症性肠病有疗效
- 下一篇:关于SNP检测方法的介绍
- 发表评论
-
- 最新评论 进入详细评论页>>
- 特别推荐
-

- 推荐内容
-
- 大脑GABA受体可作为抑郁症和认识障
抑郁症是一种复杂的疾病,与大脑功能和机制的多种差异有关。...
- 老年性痴呆,可不仅仅是记性差!
第一大征兆:记忆力衰退 记忆力衰退,尤其是对近期事物的遗忘...
- 《自然》:阿尔兹海默传染假说新
要说哪种疾病最可怕,奇点糕觉得还是 阿尔兹海默 病(Alzheimer...
- PNAS:一晚上不睡竟然会增加患阿尔
仅仅经历一晚上没有 睡 觉就会让人们大脑中淀粉样蛋白快速明...
- 保护大脑的“力量”抵御阿尔茨海
有史以来第一次,非传染性慢性病(癌症、心脏病、糖尿病和痴呆...
- 细菌真地也会在我们的大脑中安家
我们知道肠道中的 细菌 群体对我们的健康有很大的影响。这些...
- 大脑GABA受体可作为抑郁症和认识障