|
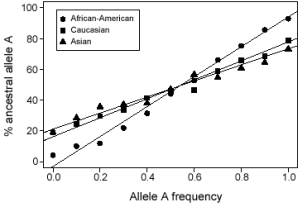
同这些理论预测一致的是,以前的一组多人种样本内小量的SNP位点研究的结果也显示了等位基因频率与祖先状态(ancestral state)的正相关性。我们通过检查3大类人种群中大量的SNP位点进一步深化了这个结果。我们将黑猩猩和大猩猩,也即祖先的等位基因分布作为非裔美国人、高加索人和亚洲人中SNP等位基因频率的功能作图,我们发现在每个例子中都存在很强的正相关性;SNP等位基因的频率越高,则祖先等位基因(ancestral allele) 的比例越高(图3) 。非裔美国人曲线的斜率是 0.97, 表明祖先状态的和等位基因频率是接近一比一的相关性,这同理论上的预测是接近一致的。相对应的高加索人和亚洲人各自为0.62和0.52。这表明这两种人群中,祖先等位基因不总是最频繁;在这两种人群中,更新的等位基因更频繁的情况约占20%。这些数据同出现在高加索人和亚洲人群的人口统计相关信息(如人口瓶颈及扩张)和现今一些选择性事件(如地区适应性)一致。这也同50,000年前人类走出非洲,迁移到欧洲和亚洲的假设是一致的。
图3在3种种群中有功能的等位基因频率的祖先等位基因的百分率
这些研究提出了同时分析14,548个SNPs,不同于采用位点特异性PCR或自动化的原理及数据。这个技术易于进行连锁分析,快速确定等位基因频率, 发现其他种群的AIMs连锁分析,确定肿瘤的DNA拷贝数变化。我们可很容易通过WGSA对更大量的(如>100,000)SNP进行分型。基因芯片的技术改进使我们可以在每张芯片上合成更多的SNP,在样本制备技术上的提高可以对更复杂的片段进行基因分型。我们的方法不仅可以对大量的SNP进行测量,也可以测量其他复杂的机体。这个技术可以测定超过100,000个SNP, 它将被用于建立haplogype maps ,及全基因组的连锁不平衡性的图谱定位。利用这些工具,在进行基因研究时可揭示复杂人类基因表型的分子基础,从而更好地理解我们的物种进化历史。
方法:
芯片设计。为了尽可能的在最小数量的芯片上进行尽可能多的SNPs的分型,我们建立了新的生化方法和用芯片进行SNP的分析,这种方法可以取代那些通过扩增来预测SNP的方法。人类基因组测序草图的完成可指导进行总基因组DNA insilico消化后, 确认所需大小的片段和预测在这些片段上的SNP位点的可能。我们排除了含有重复性序列的片段;这些片段中的SNP在TSC中大概占25-30%。我们合成了一系列的11张SNP芯片,表达了来自3种不同的基因组片段( EcoRI,BglII and XbaI)的共71.931种独特的SNP位点。每个位点设计了总计56种探针。对于每个SNP位点,4类探针(25-mer)被合成,沿着含有SNP序列的双链含概了7个位点,SNP所在的位点(位于中心),即为0位点,其他分别定为–4,-2,+1.+3,+4。正义链和反义链都设计了相应的探针,并且这两条链都分别设计7个位点的探针—— 对于2个SNP等位基因(A、B)分别设计了—个完全配对探针(PM, perfect match)和中心的单个碱基不配对的不配对的探针(MM, mismatch)。这四类探针组成一个probe quartet。将每个SNP的探针数从56个降低到40个而不降低检测的准确性是可以做到的 ( http.//www. Affymetrix.com /products /arrays/specific/10k.affx )。计算检测信号的运算公式,(PM-MM)/(PM+MM) 来判定检测信号的特异性,并使用基因分型的统计进行检测值的过滤。约9%SNP其分辨值< 0.03,将在分类前被过滤,导致的因素有很多:包括人类基因组测序草图中的错误、不易扩增带2级结构的位点,或者与其他基因序列存在交叉杂交。
DNA样本的准备。所有的样本DNA包括24个个体选自PDI-24(polymorphism discovery pane),6个不相关的个体选自于CEPH( Centre Erude Polymorphism Human),20个非裔美国人、20个亚洲人、38个高加索人选自于TSC 等位基因频率库,黑猩猩(NA03448A)和大猩猩(NF05251B),所有均通过Coriell Institute作医疗研究,这项研究是National Institute of General Medical Science Human Genetic Mutant Cell Repository中的一部分。
Target preparation
总共用250ng基因组DNA,用20单位的限制性内切酶EcoRI、BglII 或者 XbaI ( New England Biolabs(NEB)) 在37度消化4小时。在75度热失活20分钟,消化后的DNA同0.25 μM 的接头和DNA连接酶在标准连接缓冲液(NEB)中16度共同孵育4小时。然后在95度孵育5分钟热激活酶。通过扩增连接了接头的DNA来扩增靶目标,PCR缓冲液II(Perkin Elmer)包含2.5 mM MgCl2 、250μM dNTP、0.5μM的引物、50单位的Taq酶( Perkin Elmer)。PCR循环如下:95度10分钟,接着58度15秒,72度15秒,接下来95度10秒,58度15秒,72度15秒共20个循环,再接下来95度20秒,55度15秒,72度15秒共计25个循环,最后55度15秒,72度15秒。在72度延伸7分钟。这些扩增产物用YM30柱子(Microcon)在3,000*g离心6分钟进行浓缩。柱子用400μl水洗涤2次,离心14,000*g,然后颠倒(inverted),3分钟离心3,000*g 用干净管子收集。样本再用0.045单位的DNase (Affymetrix)和0.5单位的小牛小肠的磷酸酶 (Gibco ) 在RE缓冲液no.4(NEB)在37度温育30分钟。酶95度灭活15分钟。样本用15-20单位的末端转移酶标记,18μM 生物素标记的ddATP(NEN)在TdT缓冲液37度 4个小时。在热失火95度10分钟后,样品被注入芯片中,遵照操作指南(Affymetrix)杂交过夜。用0.6x SSPET 洗液在洗涤工作站中洗涤芯片,接着3步染色步骤(省略)。最后进行扫描芯片(Affymetrix)。
重复性和准确性
重复性是通过确定38个样本的基因分型的一致性来确认。38个样本的基因型检出率的百分率为91.1%到97.9%(见table 1)。平均的genotype 检出率 是95.8%± 1.2%( 平均± s.d.),证实高度的重复性。我们研究了多种酶消化片段的SNP并用2张或者更多的芯片验证。205个SNP合成于2张或者更多张芯片,分析不同的酶片段,30个个体的基因型结果的一致性为99.5%。
我们用2种方法来确定了基因型数据的准确性: 直接的,用其他技术来分析基因型;间接的,通过计算家族中孟德尔遗传错误率。均证实基因芯片有较好的重复性和准确性。
等位基因频率的确定
在3种人群的每种14,548个SNP检出值中, 我们提供每个人群中至少75%的个体中测出的SNP位点(13,647 个SNP)。比较一组高加索人(20人)同另外一组高加索人(28人)的等位基因频率,显示了他们之间的高度相关性(R2= 0.96),表明40个染色体样本提供了在这种人类种群中的SNP等位频率的稳定的评估。
标记分布(Marker distribution)
所有的SNP信息均可在TSC中找到。SNP的间距的平均值和中点值分别为173kb和80.8kb。所有这些标记中,5,058个SNP位点被定位在50kb 或者更短的间距。3,868个定位在25kb或者更短的间距。
数据来源
来自于108个个体所提供14,548个 SNP分型的信息(包括从等位基因研究中的60个个体的所得到的信息)以及黑猩猩和大猩猩的基因型,所有信息均可在 http://ncbi.nlm .gov/SNP/under the handle”AFFY.” 中查询到。
(责任编辑:泉水) |
